MRC Prion Unit at UCL, Institute of Prion Diseases
Courtauld Building, 33 Cleveland Street, London, W1W 7FF (Telephone: +44 (0) 207 679 5147)
 Close
Close

Courtauld Building, 33 Cleveland Street, London, W1W 7FF (Telephone: +44 (0) 207 679 5147)
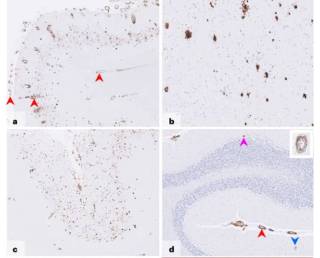
Iatrogenic Alzheimer’s disease in recipients of cadaveric pituitary-derived growth hormoneWe describe a new type of Alzheimer’s disease – iatrogenic Alzheimer’s disease – occurring after treatment with cadaveric human growth hormone many decades earlier.